Reconstruct and analyse multi-omics networks with carmon
Alessio Albanese
Source:vignettes/carmon.Rmd
carmon.RmdIntroduction
Copula-Aided Reconstruction of Multi-Omics Networks () is an R
package empowered by the use of copulas as a statistical tool for the
reconstruction of multi-omics networks. The use of copulas as a
statistical tool allows to transfer raw non-normalized omics data to the
normal realm. In this way, the package is able to harness the full
statistical power of the information contained in each omics layer of a
multi-omics data set. At the same time, the transfer of the multi-omics
layer to the normal realm allows the application of network
reconstruction models that assume normal distribution of the data.
Needless to say, this is an assumption rarely met by omics data, hence a
further advantage of the use of copulas. Here, we first cover the
formatting of the input data to provide to . Then, we show how to easily
build and analyse a multi-omics network with carmon. In the remaining
sections, we explore more advanced and customized uses of the package,
as well as what actually happens under the hood when the main function
carmon() is used.
Installation
You can install from Bioconductor with:
if (!require("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("carmon")Alternatively, you can install the development version from GitHub. First, make sure to install devtools with:
if (!require("devtools")) {
install.packages("devtools")
}You can then install the development version with:
devtools::install_github("DrQuestion/carmon")Input multi-omics data set-up
We first attach the package.
We can now explain the data format. Carmon prefers the input
multi-omics data to be arranged in a named R list. Each element of the
list should be the data set of one of the omics layers, and it should be
preferably named after the omics type it contains. In the package there
are several examples of multi-omics data sets of increasing sizes, which
you can see with help(multi_omics). Here we use
multi_omics_small.
data(multi_omics_small)
str(multi_omics_small)
#> List of 2
#> $ rnaseq : num [1:30, 1:14] 1023 815 1047 725 1217 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : chr [1:30] "BXD100" "BXD101" "BXD73b" "BXD44" ...
#> .. ..$ : chr [1:14] "Cirbp" "Hspa5" "P4ha1" "Spred1" ...
#> $ metabolomics: num [1:30, 1:5] 66.1 50.2 56.3 58.9 61 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : chr [1:30] "BXD100" "BXD101" "BXD73b" "BXD44" ...
#> .. ..$ : chr [1:5] "Phe" "Trp" "Putrescine" "PC aa C36:3" ...You can see that this list has two elements, one containing RNA-seq
gene counts and named “rnaseq” (multi_omics_small$rnaseq)
and the other containing metabolites concentrations and named
“metabolomics” (multi_omics_small$metabolomics). Each layer
has 30 observations, the first one measuring the expression of 14 genes,
the second one the concentration of 5 metabolites. Please notice that
carmon expects non-normalized data, to harness at the best the
statistical behavior of omics data with copula.
To recreate such an object on R, first it is necessary to make sure that the observations/samples/individuals from which the data set has been measured are in the same order for all the input omics data sets and are consistent in where they are placed for all data sets, either all across the rows or all across the column. For example, in our toy data set, you can see how all the observations are in the same order and distributed along the rows for both the transcriptomic and the metabolomic data set. After your separate data sets are properly formatted, you can assemble them in a single list as follows:
# Let's build two separate data sets just for the sake of our example:
layer_1 <- multi_omics_small$rnaseq
layer_2 <- multi_omics_small$metabolomics
# This shows that observations are on the rows for both data sets:
dim(layer_1) # [1] 30 14
dim(layer_2) # [1] 30 5
# This shows that the observations are all in the same order in both data sets:
all(rownames(layer_1) == rownames(layer_2)) # TRUE
# We can now build our R list as follows:
multi_omics_small <- list("rnaseq" = layer_1, "metabolomics" = layer_2)Using MultiAssayExperiment objects or single unified
data sets as input
It is also possible to provide carmon() with a MultiAssayExperiment
or a single unified multi-omics data set. In the latter, it is necessary
to specify the argument p, a vector with the number of
features of each layer, in the same order as the layers are assembled in
the data set. Moreover, with both these use cases, we strongly recommend
using the omics argument to specify which omics type is
measured by each layer. To see which omics types carmon()
is tailored for, together with their accepted synonyms and assumed
statistical behavior, see which_omics().
Carmon to perform copula-aided reconstruction, analysis and plot of a multi-omics network
The carmon package covers multiple functional steps of network reconstruction and analysis from multi-omics data. To perform a full, default run of carmon, it is necessary to run:
c_obj <- carmon(multi_omics_small)
#> Checking sample-matching, formatting the data,
#> and other formalities....
#> Done!
#>
#> ****************Beginning copulization****************
#> Copulizing layer 1 of 2 (rnaseq)
#> Copulizing layer 2 of 2 (metabolomics)
#> ****************Copulization complete*****************
#> ***********Beginning network reconstruction***********
#> Reconstructing network with coglasso, selecting the optimal one with xestars....
#> Optimal network selected!
#> ***********Network reconstruction complete************
#> **************Beginning network analysis**************
#> Computing centrality measures....
#> Centrality measures computed!
#> Compiling the report....
#> Report compiled!
#> **************Network analysis complete***************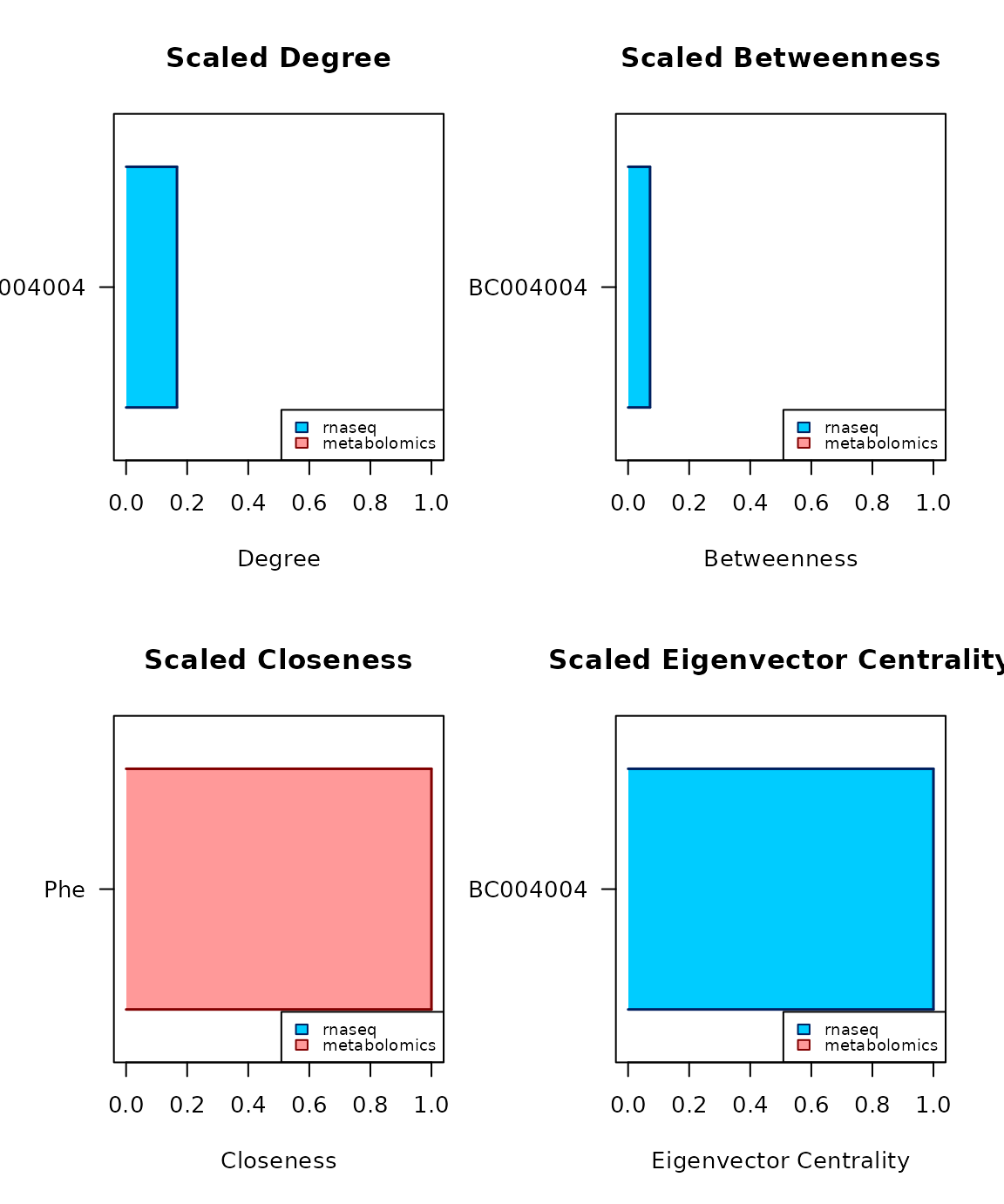
Under the hood, there are several processes happening. In order: the
check-ups and formatting of the input data; the copula-based transfer of
non-normal omics data to the normal realm; the reconstruction and
selection of a multi-omics network; a centrality consensus analysis to
identify key components of the network; and the plotting of the
multi-omics network (enriched by the information discovered during the
centrality analysis) and of the results of the centrality analysis. Most
of these steps can be customized within the main wrapper function, using
specific arguments. We will see how in the next sections, but first
let’s access the results of the centrality consensus analysis with the
function centrality_report():
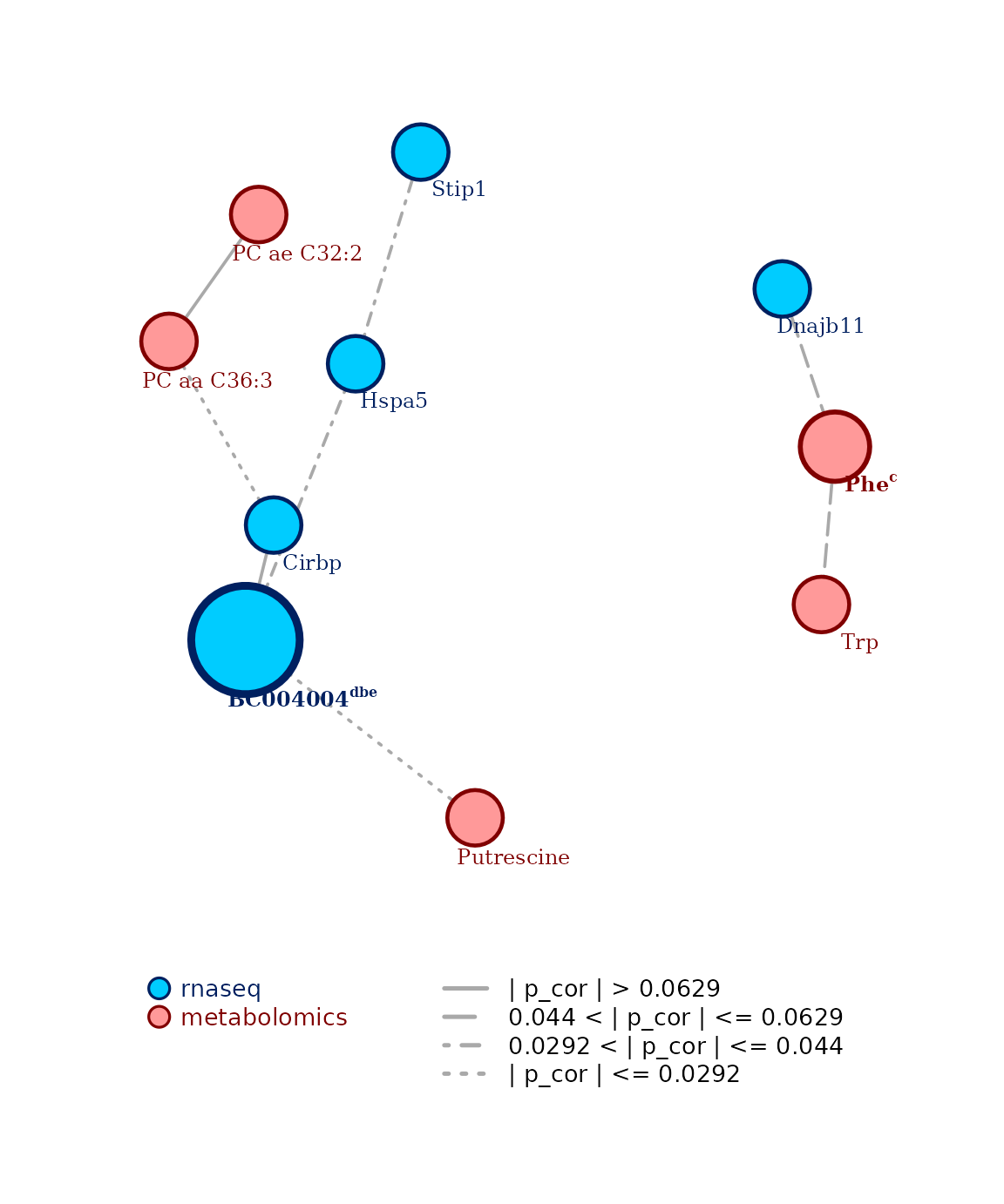
centrality_report(c_obj)
#> candidate degree betweenness closeness eigenvector central for
#> BC004004 BC004004 0.1667* 0.0719* 0.6 1* dbe
#> Phe Phe 0.1111 0.0065 1* 0 cCopula-based transfer to the normal realm
Carmon harnesses the raw statistical behavior of the non-normally distributed omics data to perform a proper transfer to the normal realm. The statistical tool the package uses for this task are Gaussian copulas. The first ingredient to perform this transition with copulas is the choice of what statistical behavior best describes your omics data, by deciding which marginal distribution is followed by each omics layer.
The most basic approach to this is to choose the empirical distribution, with which we take the data as they are, without making any assumption. The approach carmon prefers, when possible, is to assume that the data follow specific parametric statistical distributions. With this approach, we use the omics data to estimate the parameters of such distribution, allowing to find the distribution of the chosen family that best describes the omics data.
With specific types of omics data, over the years, the scientific
community has settled on specific distributional assumptions. For
example, RNA-seq data, together with other omics types that generate
count-based measures, are generally assumed to follow a Negative
Binomial distribution. Carmon takes several of this known distributional
assumptions and implements them as a default to tailor its treatment of
those specific omics types. For all those omics types that have no known
assumption yet, or those for which we have not tailored a default
behavior yet, carmon chooses the first approach, the one based on the
assumption-free empirical distribution. To see for which omics types we
tailored a default behavior, please use the function
which_omics(). Be aware that carmon is built for being
upgraded, so soon there will be new omics types for which we implement a
tailored default behavior.
which_omics()
#> "rna-seq", also as "rnaseq", "rna", "gene counts", "transcriptomics"
#> is modeled by default as count data with a negative-binomial marginal.
#>
#> "mirna-seq", also as "mirnaseq", "microrna-seq", "micrornaseq", "mirna",
#> "microrna"
#> is modeled by default as count data with a negative-binomial marginal.
#>
#> "proteomics", also as "protein fragments", "protein counts"
#> is modeled by default as count data with a negative-binomial marginal.
#>
#> "metabolomics", also as "lc-ms", "gc-ms", "ms"
#> is modeled by default as positive continuous data with a log-normal
#> marginal.
#>
#> Anything else is modeled with the empirical marginal.These are also the terms you can use to name the elements of the
input R list if you want to trigger the default behavior for the
corresponding layers. For example, you could see that the two elements
in the input list multi_omics_small are
"rnaseq", which will trigger the default assumption of
Negative Binomial distribution to model gene counts, and
"metabolomics", which will trigger by default the
log-normal distribution to describe the metabolites’ concentrations.
If this is the default behavior for such omics types, but you know
that your particular data set follows a different statistical
distribution, you can run the function which_marginals().
This function displays which are the statistical distributions that
carmon is currently compatible with.
which_marginals()
#> "e" or "empirical" for using the empirical marginal distribution;
#> "n" or "normal" for using the normal marginal distribution;
#> "ln" or "lognormal" for using the log-normal marginal distribution;
#> "nb" or "negative binomial" for using the log-normal marginal distribution.This information can be used to customize the default behavior of
carmon with the copula-based transition to normal data. The two
arguments to the main wrapping function carmon() that you
can use for this are:
-
omics: to be provided as a vector of characters, by default it is unnecessary, as the input list will have the omics types in the name given to each element. We recommend its use especially when for some reason the elements are not or cannot be named. An example of it could be when two layers in the list measure the same omics type, so you may want to explicitate it as an input with this argument. This is how you can use it in our exampleomics = c("rnaseq", "metabolomics"). -
marginals: also a vector of characters, it overrides the default behavior for the input omics types. It is also possible to have a mixed case, in which for some omics layers you want to trigger the default, and for others you want to customize it. If you know that for your first layer you would rather use the assumption-free empirical distribution, the normal for the second, and you prefer normal behavior for the third, this is the form that the argument could take:marginals = c("e", "n", 0), requesting default behavior with the0.
Network reconstruction and selection
Once the omics layers have been transferred to the normal realm,
carmon() proceeds to build the multi-omics network. For this task the
package implements multiple network reconstruction strategies, of which
one is chosen. By defualt, the package chooses collaborative
graphical lasso (coglasso) (Albanese,
Kohlen and Behrouzi, 2024) as its engine to reconstruct the
multi-omics network, as the method was specifically developed for the
multi-omics network reconstruction scenario. carmon also implements
three classic alternatives to the reconstruction of networks:
graphical lasso (Friedman, Hastie and
Tibshirani, 2008), neighborhood selection (Meinshausen and Buhlmann, 2006), and Pearson’s
correlation networks. The network reconstruction strategy can be
personalized with the argument net_method, for example
net_method = "coglasso", while for choosing one of the
other ones, respectively, you would need "glasso",
"mb", or "correlation".
Apart from one specific use case, each of these methods explores
network reconstruction among a range of hyperparameters, usually
resulting in a different network for each different value (or
combination of values) these hyperparameters can take. This requires
specific model selection procedures that can pinpoint the best network
among the ones that have been built. Luckily, for each network
reconstruction method it implements, carmon() also comes
with its tailored model selection procedures. For coglasso, for
example, it is possible to choose among three possible strategies. One
of them is the extended Bayesian Information Criterion
(eBIC). The other two are different versions of an algorithm
based on StARS (Liu, Roeder and Wasserman, 2010),
extended to the coglasso case in which three different
hyperparameters need to be tuned, and described in (Albanese, Kohlen and Behrouzi, 2024) as XStARS.
The two different versions implemented are indeed the original
XStARS and the more efficient but slightly less thorough
version XEStARS. By default, for coglasso as a network
reconstruction method carmon() uses XEStARS. To
customize the model selection procedure, you can use the
sel_method parameter. For example, if instead of the
default you prefer the more thorough XStARS, you can set
sel_method = "xstars", with the default being
"xestars" and "ebic" being the alternative for
using eBIC. For every other network reconstruction procedure,
as they are all based on a single hyperparameter, carmon()
uses by default the one-dimensional StARS, but more options are
available. See them all in help(carmon), under the
description of the sel_method argument.
All of the methods mentioned above, either for network reconstruction
or network selection, can be further tweaked by giving specific
arguments. For example, if using coglasso for reconstruction,
and wanting to investigate the hyperparameter space more thoroughly than
the default, one could specify the coglasso-specific parameters
with a higher number, say nlambda_w = 20 (normally only 8
values are explored), nlambda_b = 20 (only 8 values
normally), and nc = 10 (only 5 normally). To see more about
these and other arguments one can tweak about coglasso network
reconstruction and selection, please see ?coglasso::bs. For
the other network reconstruction methods, please see
?huge::huge.glasso for graphical lasso,
?huge::huge.mb for neighborhood selection, and
?huge::huge.ct for Pearson’s correlation, and see
?huge::huge.select for their associated model selection
procedures.
One specific run mode of carmon() does not require model
selection, because the user is directly setting a value required
hyperparameter. It is possible when using
net_method = "correlation" and setting one between
cor_cutoff and cor_quant. The first determines
the threshold value of absolute Pearson’s correlation below which the a
connection should be excluded, while the second determines the quantile
of top connections (per absolute value) that need to be included in the
network. For example, setting cor_quant = 0.05 will include
in the network only the top 5% connections based on the absolute
Pearson’s correlation value.
Centrality consensus analysis
Once carmon() selects a final network, it proceeds with
performing a centrality consensus analysis. In this step, four different
centrality measures are computed for the nodes of the network, with the
idea that the larger the consensus among the different measures about a
specific omics feature, the more likely this feature is to be important
to the studied biological phenomenon. The four measures employed are
degree centrality, betweenness centrality,
closeness centrality, and eigenvector centrality. By
default all the four of them are used for the consensus, but this can be
personalized with the argument c_measures, using the first
letter of each measure you want to employ. For example, to use only the
first three measures, you can set c_measures = "dbc", while
the default would be "dbce".
Determining the size of the consensus
It is also possible to personalize how many omics features will be
included in the consensus with two arguments:
max_candidates_c_measures, and
quantile_c_measures. Both arguments determine the number of
top candidates selected by each measure separately, before
finding the consensus among the measures. The first does it with the
absolute value of candidates, the other with a determined quantile of
top candidates. The default values for these two arguments are
max_candidates_c_measures = 20 and
quantile_c_measures = 0.05, and the smallest between the
two will be used. The same principle holds true when the user gives
values for both arguments, but when only one of the two is given that
one only determines the amount of candidates for the consensus.
Please note that the centrality analysis is the first optional step,
and it can be turned off by setting the argument to
carmon() analyse = FALSE.
Plot of network and of centrality analysis results
The final functional step of carmon() is the plotting
module. carmon() can generate two plots, depending on
whether the centrality analysis is carried on or not. The first one it
will show when the analysis is performed is the measures of centrality
for the top candidates, separately for each measure. This means that, by
default, four panels will be generated when all the four measures found
central nodes, but when a measure fails to find central nodes, then its
panel will not be displayed.
The second plot generated is the selected multi-omics network,
enriched by the results of the centrality analysis when this one is
performed. When an omics feature is found to be central according to a
measure, the first letter of the measure is added at the end of the
displayed node label, and the node itself is plotted larger. This also
means that the larger the consensus, the larger the node will be. At the
bottom of the plot, a legend shows the color coding for the different
omics types of the network and the different edge types for the
different edge strengths, depicting how strong the connection is. The
multi-omics network plot can be personalized with three different
arguments to carmon():
-
plot_hot_nodescan be set toFALSEto turn off the enrichment of the network based on the centrality analysis, and it is turned on by default. -
plot_node_labelscan be set toFALSEto hide the node labels from the plot, which is particularly useful for larger networks. Whenplot_hot_nodesisTRUE(default behavior) andplot_node_labelsisFALSE, only the central nodes’ labels are displayed in the plot. -
hide_isolatedhides the unconnected nodes from the plot of the multi-omics network,TRUEby default.
Please note that also the plotting module can be turned off by
setting the argument to carmon()
plot = FALSE.
Custom run of carmon()
In this run, we customize the default behavior of
carmon() with the information provided above, this time
reconstructing the network from the largest data set provided by the
package, multi_omics. We will will use alternative
marginals for the RNA-seq layer, keeping the default for the metabolomic
layer. We will use the omics features in the normal-realm to build a
network with Pearson’s correlation, selecting only the top 5%
connections. We will perform the consensus centrality analysis with all
four measures, but we will select the top 2% for each measure to select
the candidates for consensus. Moreover, we will hide the name of those
nodes that are not found to be central. Finally, we will inspect the
result of the consensus centrality analysis.
data(multi_omics)
str(multi_omics) # 162 genes, 76 metabolites, for 30 observations
#> List of 2
#> $ rnaseq : num [1:30, 1:162] 407 54 148 269 76 91 500 482 65 201 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : chr [1:30] "BXD100" "BXD101" "BXD73b" "BXD44" ...
#> .. ..$ : chr [1:162] "Plin4" "Arc" "Egr2" "Tekt4" ...
#> $ metabolomics: num [1:30, 1:76] 317 208 255 282 362 ...
#> ..- attr(*, "dimnames")=List of 2
#> .. ..$ : chr [1:30] "BXD100" "BXD101" "BXD73b" "BXD44" ...
#> .. ..$ : chr [1:76] "Ala" "Arg" "Asn" "Cit" ...
c_obj <- carmon(multi_omics,
marginals = c("e", 0), # Sets the empirical marginal distribution
# for the first layer, and the default
# distribution for the second
net_method = "correlation", # Selecting Pearson's correlation
cor_quant = 0.05, # Selecting the top 5% of connections
quantile_c_measures = 0.02, # Top 2% of the candidates
# to determine consensus of centrality
plot_node_labels = FALSE
)
#> Checking sample-matching, formatting the data,
#> and other formalities....
#> Done!
#>
#> ****************Beginning copulization****************
#> Copulizing layer 1 of 2 (rnaseq)
#> Copulizing layer 2 of 2 (metabolomics)
#> ****************Copulization complete*****************
#> ***********Beginning network reconstruction***********
#> Reconstructing network with Pearson's correlation....
#> Correlation cutoff is 0.64002200550331
#> Network reconstructed!
#> ***********Network reconstruction complete************
#> **************Beginning network analysis**************
#> Computing centrality measures....
#> Centrality measures computed!
#> Compiling the report....
#> Report compiled!
#> **************Network analysis complete***************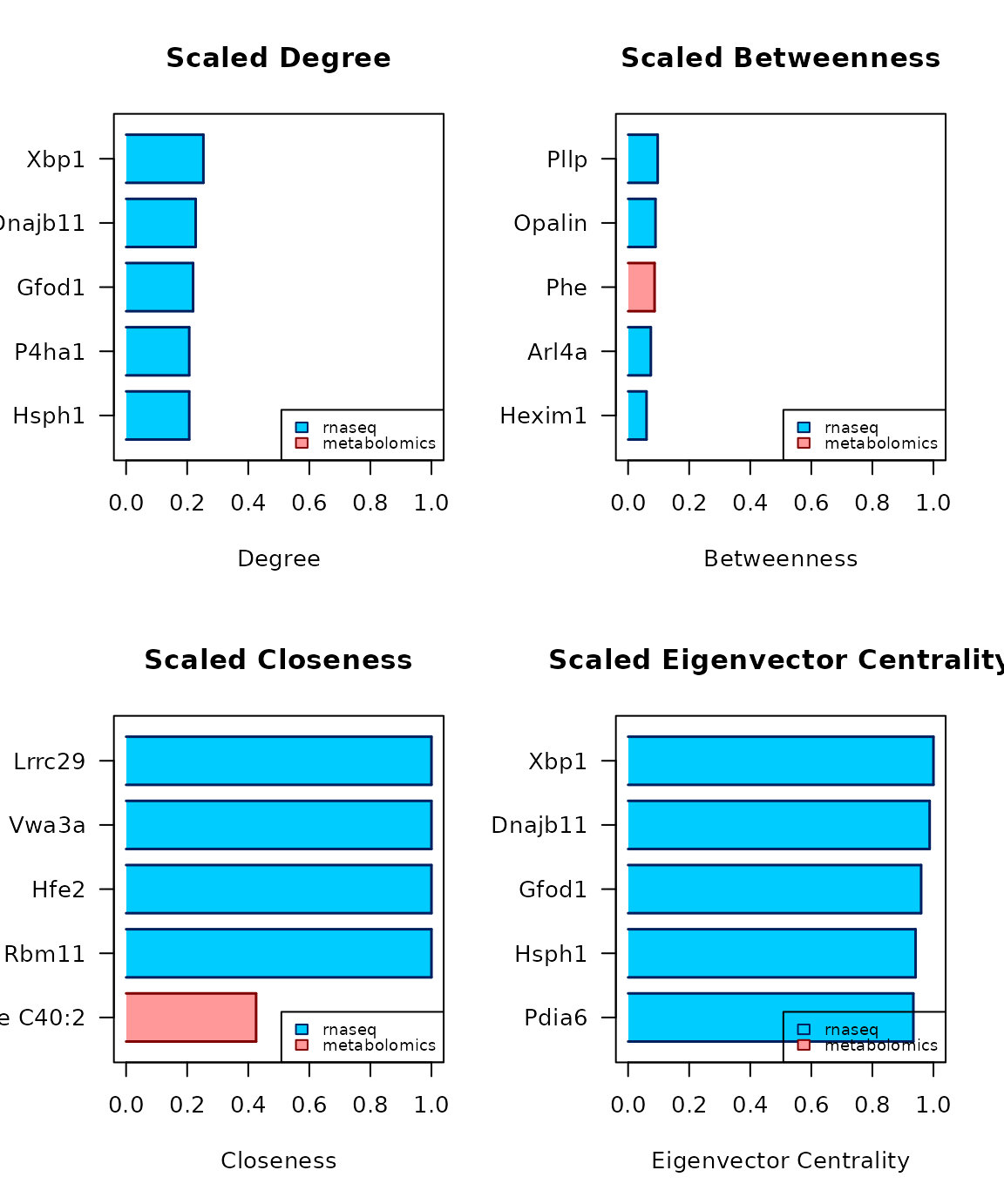
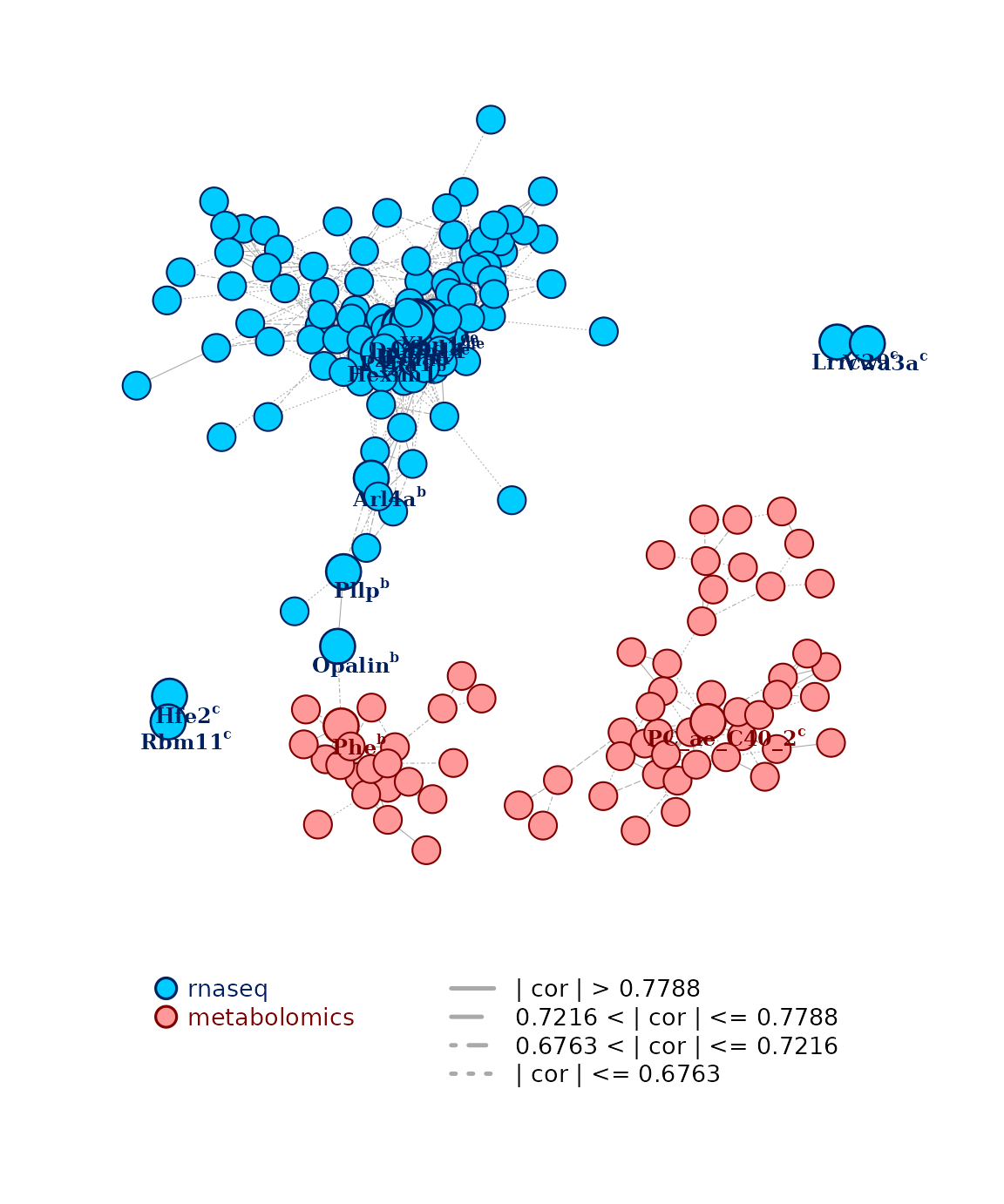
centrality_report(c_obj)
#> candidate degree betweenness closeness eigenvector central for
#> Xbp1 Xbp1 0.2532* 0.0226 0.4072 1* de
#> Dnajb11 Dnajb11 0.2278* 0.0076 0.3965 0.9875* de
#> Gfod1 Gfod1 0.2194* 0.0062 0.3908 0.9597* de
#> Hsph1 Hsph1 0.2068* 0.0045 0.3886 0.9415* de
#> Pdia6 Pdia6 0.1983 0.0032 0.3853 0.9342* e
#> P4ha1 P4ha1 0.2068* 0.0054 0.3853 0.9007 d
#> Hexim1 Hexim1 0.1899 0.0604* 0.4159 0.8308 b
#> Lrrc29 Lrrc29 0.0042 0 1* 0 c
#> Vwa3a Vwa3a 0.0042 0 1* 0 c
#> Hfe2 Hfe2 0.0042 0 1* 0 c
#> Rbm11 Rbm11 0.0042 0 1* 0 c
#> PC ae C40:2 PC ae C40:2 0.0717 0.0107 0.4257* 0 c
#> Arl4a Arl4a 0.038 0.0745* 0.34 0.0542 b
#> Pllp Pllp 0.0211 0.0969* 0.2804 0.0025 b
#> Opalin Opalin 0.0084 0.0897* 0.2361 1e-04 b
#> Phe Phe 0.0422 0.0867* 0.2033 0 bReferences
Albanese, A., Kohlen, W., & Behrouzi, P. (2024). Collaborative graphical lasso (arXiv:2403.18602). arXiv https://doi.org/10.48550/arXiv.2403.18602
Friedman, J., Hastie, T., & Tibshirani, R. (2008). Sparse inverse covariance estimation with the graphical lasso. Biostatistics, 9(3), 432–441. https://doi.org/10.1093/biostatistics/kxm045
Meinshausen, N., & Buhlmann, P. (2006). High-dimensional graphs and variable selection with the Lasso. Ann. Statist., 34(3), 1436-1462. https://doi.org/10.1214/009053606000000281
Liu, H., Roeder, K., & Wasserman, L. (2010). Stability Approach to Regularization Selection (StARS) for High Dimensional Graphical Models (arXiv:1006.3316). arXiv https://doi.org/10.48550/arXiv.1006.3316
Session Info
sessionInfo()
#> R version 4.5.2 (2025-10-31)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] carmon_0.99.0 BiocStyle_2.38.0
#>
#> loaded via a namespace (and not attached):
#> [1] SummarizedExperiment_1.40.0 gtable_0.3.6
#> [3] xfun_0.54 bslib_0.9.0
#> [5] ggplot2_4.0.0 Biobase_2.70.0
#> [7] lattice_0.22-7 vctrs_0.6.5
#> [9] tools_4.5.2 generics_0.1.4
#> [11] stats4_4.5.2 parallel_4.5.2
#> [13] tibble_3.3.0 pkgconfig_2.0.3
#> [15] BiocBaseUtils_1.12.0 Matrix_1.7-4
#> [17] RColorBrewer_1.1-3 S7_0.2.0
#> [19] desc_1.4.3 S4Vectors_0.48.0
#> [21] lifecycle_1.0.4 compiler_4.5.2
#> [23] farver_2.1.2 textshaping_1.0.4
#> [25] DESeq2_1.50.0 Seqinfo_1.0.0
#> [27] codetools_0.2-20 htmltools_0.5.8.1
#> [29] sass_0.4.10 yaml_2.3.10
#> [31] pkgdown_2.1.3 pillar_1.11.1
#> [33] jquerylib_0.1.4 BiocParallel_1.44.0
#> [35] DelayedArray_0.36.0 cachem_1.1.0
#> [37] abind_1.4-8 locfit_1.5-9.12
#> [39] tidyselect_1.2.1 digest_0.6.37
#> [41] dplyr_1.1.4 bookdown_0.45
#> [43] fastmap_1.2.0 grid_4.5.2
#> [45] cli_3.6.5 SparseArray_1.10.1
#> [47] magrittr_2.0.4 S4Arrays_1.10.0
#> [49] withr_3.0.2 scales_1.4.0
#> [51] rmarkdown_2.30 XVector_0.50.0
#> [53] matrixStats_1.5.0 igraph_2.2.1
#> [55] ragg_1.5.0 evaluate_1.0.5
#> [57] knitr_1.50 GenomicRanges_1.62.0
#> [59] IRanges_2.44.0 MultiAssayExperiment_1.36.0
#> [61] rlang_1.1.6 Rcpp_1.1.0
#> [63] glue_1.8.0 BiocManager_1.30.26
#> [65] BiocGenerics_0.56.0 jsonlite_2.0.0
#> [67] R6_2.6.1 MatrixGenerics_1.22.0
#> [69] systemfonts_1.3.1 fs_1.6.6
#> [71] coglasso_1.1.0